
| Hội chứng Klinefelter | |
|---|---|
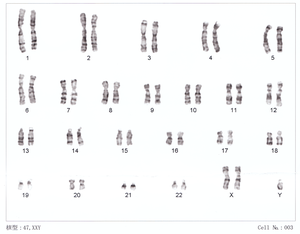
| 47,XXY | |
| Chuyên khoa | Di truyền học y khoa |
| ICD-10 | Q98.0-Q98.4 |
| ICD-9-CM | 758.7 |
| eMedicine | ped/1252 |
| Patient UK | Hội chứng Klinefelter |
| MeSH | D007713 |
Hội chứng Klinefelter (đọc là Clai-phen-tơ) là tình trạng rối loạn nhiễm sắc thể ở nam giới, nơi mà người bị ảnh hưởng có một cặp nhiễm sắc thể X thay vì chỉ một. Hội chứng này liên quan đến nhiều nguy cơ về sức khỏe và được đặt tên theo bác sĩ Harry Klinefelter, người đầu tiên mô tả tình trạng này vào năm 1942 tại Bệnh viện Đa khoa Massachusetts, Boston.
Nguyên nhân gây bệnh
Kiểu sắp xếp nhiễm sắc thể XXY là biến thể di truyền phổ biến nhất ở nam giới, xảy ra khoảng 1 trong mỗi 500 đến 1000 trẻ nam khi sinh tại Hoa Kỳ, với khoảng 3000 trẻ mắc mới mỗi năm. Do có thêm một nhiễm sắc thể, người bị ảnh hưởng thường được gọi là 'Nam XXY' hoặc 'Nam 47,XXY' hơn là 'mắc hội chứng Klinefelter'.
Ở động vật có vú với hơn một nhiễm sắc thể X, chỉ có một nhiễm sắc thể X hoạt động, trong khi các nhiễm sắc thể X khác bị bất hoạt và hầu hết các gen trên chúng không biểu hiện. Điều này xảy ra cả ở nam XXY và nữ XX. Tuy nhiên, một số ít gen trên nhiễm sắc thể X bất hoạt vẫn biểu hiện, được gọi là các gen trong vùng giả nhiễm sắc thể. Các gen này có thể là nguyên nhân của các triệu chứng liên quan đến hội chứng Klinefelter ở nam XXY.
Báo cáo đầu tiên về người đàn ông có kiểu nhân 47,XXY được công bố bởi Patricia A. Jacobs và Bác sĩ J.A. Strong tại Bệnh viện Đa khoa Miền Tây ở Edinburgh, Scotland vào năm 1959. Người đàn ông 24 tuổi này biểu hiện hội chứng Klinefelter. Bác sĩ Jacobs đã miêu tả khám phá về dị bội nhiễm sắc thể ở người và động vật có vú trong bài diễn văn Giải thưởng tưởng niệm William Allan năm 1981.
Khoảng 50-60% trường hợp không phân li nhiễm sắc thể xảy ra ở mẹ (75% do lỗi phân bào giảm nhiễm I). Các trường hợp còn lại xảy ra do không phân li ở người cha. Kiểu nhân thường gặp nhất là 47,XXY (80-90%); thể khảm 46,XY/47,XXY gặp khoảng 10%. Các thể khác như 48,XXYY, 48,XXXY, 49,XXXYY, và 49,XXXXY rất hiếm. Thể khảm 46,XY/47,XXY có thể xảy ra ở hợp tử 46,XY hoặc 47,XXY do rối loạn phân bào sau thụ tinh.
Sinh lý bệnh học
Thêm một hoặc nhiều nhiễm sắc thể X hoặc Y gây ra các bất thường về thể chất và nhận thức. Mức độ bất thường kiểu hình và chậm phát triển trí tuệ liên quan đến số nhiễm sắc thể X thêm vào. Số lượng nhiễm sắc thể X tăng làm ảnh hưởng phát triển thể chất và nhận thức, gây bất thường xương và tim mạch, rối loạn sinh dục, thiểu sản cơ quan sinh dục, và giảm chỉ số IQ khoảng 15 điểm cho mỗi nhiễm sắc thể X vượt mức bình thường.
Thêm nhiễm sắc thể giới tính, thường do lỗi không phân li trong quá trình tạo giao tử, gây suy sinh dục, vú to, và rối loạn tâm lý xã hội. Hội chứng Klinefelter là suy tinh hoàn nguyên phát, kèm theo tăng gonadotropin do thiếu phản hồi ngược từ tuyến yên. Thiếu hụt androgen gây tỉ lệ phần trên cơ thể giống người bị hoạn, lông mặt và thân mình thưa thớt, vú to, tinh hoàn và dương vật nhỏ, giảm khoái cảm tình dục, phân bố mỡ kiểu phụ nữ, giảm sức chịu đựng thực thể, loãng xương. Mất ống sinh tinh và tế bào Sertoli dẫn đến giảm inhibin B, điều hoà hormone FSH. Trục hạ đồi-yên-sinh dục thay đổi ở bệnh nhân hội chứng Klinefelter tuổi dậy thì.
Cũng có báo cáo về nguy cơ tăng các bệnh tự miễn như lupus ban đỏ hệ thống, viêm khớp dạng thấp, và hội chứng Sjögren. Điều này có thể do giảm testosterone và tăng estrogen, vì androgen có thể bảo vệ chống tự miễn, trong khi estrogen có thể thúc đẩy các rối loạn này.
Dấu hiệu và triệu chứng
Phần lớn nam giới sinh ra với hội chứng Klinefelter sống suốt đời mà không được chẩn đoán. Khi chẩn đoán, thường xảy ra ở tuổi trưởng thành, thường do thiểu năng sinh dục và vô sinh. Tỉ lệ tử vong không cao hơn nhiều so với người bình thường.
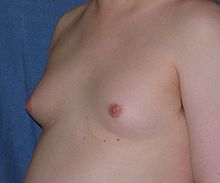
Lý do thường gặp để bệnh nhân đi khám là vô sinh và vú to. Các triệu chứng khác bao gồm mệt mỏi, yếu, rối loạn cương dương, loãng xương, rối loạn ngôn ngữ, khó khăn trong học hành, khoái cảm tình dục giảm, tự trọng kém, và các rối loạn hành vi.
Về mặt thực thể, trẻ sơ sinh và trẻ nhỏ có chiều cao, cân nặng và vòng đầu bình thường. Khoảng 25% biểu hiện lệch ngón (clinodactyly). Tốc độ phát triển chiều cao tăng vào lúc 5 tuổi, và chiều cao người lớn thường cao hơn mức trung bình. Ngoài ra, tay và chân cũng dài không tỉ lệ với cơ thể. Một số người với thể Klinefelter 49,XXXXY có vóc người thấp.
Phần lớn nam giới 47,XXY có trí thông minh bình thường, nhưng IQ bị ảnh hưởng bởi nền tảng gia đình. Sự hiện diện của nhiều nhiễm sắc thể X hơn có thể dẫn đến trí tuệ dưới mức bình thường hoặc chậm phát triển tâm thần.
Khoảng 70% bệnh nhân gặp khó khăn nhẹ về phát triển tâm thần và học hành, bao gồm học tập khó khăn, chậm nói, trí nhớ ngắn hạn kém, kỹ năng phục hồi dữ liệu giảm, khó đọc, chứng khó đọc (dyslexia), và khiếm khuyết khả năng chú ý. Các rối loạn hành vi và tâm lý cũng phổ biến, có thể do tự trọng thấp và khả năng xử lý căng thẳng giảm. Rối loạn tâm thần như lo lắng, trầm cảm, loạn thần kinh (neurosis), và loạn thần (psychosis) cũng thường gặp hơn ở nhóm này so với dân số chung.
Răng bò (taurodontism, răng hàm phình to do tuỷ nở rộng) xuất hiện ở 40% bệnh nhân, trong khi tỉ lệ này ở người XY bình thường chỉ là 1%.
Về đặc điểm giới tính, bệnh nhân có thể mất các đặc điểm giới tính thứ phát do giảm sản xuất androgen, biểu hiện bằng lông ở mặt, thân và cơ quan sinh dục thưa, giọng nói cao và phân bố mỡ kiểu phụ nữ. Khoảng 30-50% trẻ nam mắc hội chứng Klinefelter biểu hiện vú to thứ phát vào giai đoạn muộn tuổi dậy thì do tăng estradiol và tỉ lệ estradiol/testosterone cao. Nguy cơ phát triển carcinoma vú cao hơn ít nhất 20 lần so với người bình thường. Loạn sản tinh hoàn (tinh hoàn nhỏ, chắc, kích thước < 10ml) có thể có sau dậy thì. Vô sinh, vô tinh trùng có thể do teo ống sinh tinh. Tất cả bệnh nhân 47,XXY đều vô sinh; nhưng bệnh nhân thể khảm 46,XY/47,XXY có thể còn khả năng sinh sản. Họ cũng có thể tăng tần suất u tế bào mầm ngoài sinh dục như carcinoma phôi, u quái và u tế bào mầm trung thất nguyên phát.
Về rối loạn tuần hoàn và tim mạch, 55% bệnh nhân bị sa van hai lá, 20-40% bị dãn tĩnh mạch, nguy cơ loét tĩnh mạch cao gấp 10-20 lần so với người bình thường, nguy cơ huyết khối tĩnh mạch sâu và thuyên tắc phổi cũng tăng.
Xét nghiệm di truyền tế bào cho thấy kiểu nhân bất thường 47,XXY hoặc các biến thể khác. Nồng độ FSH, LH và estradiol trong máu tăng, trong khi testosterone giảm ở bệnh nhân từ 12-14 tuổi. Đáp ứng testosterone khi tiêm hCG thấp hơn bình thường. Gonadotropin niệu tăng do rối loạn chức năng tế bào Leydig. Nồng độ osteocalcin huyết thanh và tỉ lệ hydroxyl proline/creatinine tăng, phản ánh giảm tạo xương và tăng tái hấp thu xương.
Khảo sát hình ảnh cho thấy sa van hai lá qua siêu âm, giảm mật độ xương chi dưới, dính xương quay trụ, và răng bò trên X quang.
Khảo sát mô học cho thấy tinh hoàn nhỏ, chắc với hiện tượng hyalin hoá, xơ hoá và teo ống sinh tinh kèm tăng sản khu trú của các tế bào Leydig đã bị thoái hoá. Tế bào mầm giảm hoặc không có. Hiện tượng sinh tinh trùng hiếm gặp. Ở bệnh nhân thể khảm, hyalin hoá và thoái hoá tiến triển ống sinh tinh xảy ra sau dậy thì dù tinh hoàn có kích thước bình thường lúc đó. Mô học vú to cho thấy tăng sản mô gian ống. Đột biến số lượng NST dạng thể ba ở cặp NST giới tính gây hội chứng này ở trẻ trai có hai hoặc nhiều hơn 2 nhiễm sắc thể X, có thân cao không bình thường, tinh hoàn nhỏ, si đần, không có con.
Chẩn đoán trước khi sinh
Chẩn đoán trước sinh có thể thực hiện qua chọc ối và phân tích di truyền tế bào dịch ối. Điều này có thể đặt cha mẹ vào tình thế khó xử, do dự hậu tốt nhưng vẫn có khả năng bất thường kiểu hình. Chỉ rất ít trường hợp thể khảm 46,XY/47,XXY có khả năng sinh con, với nguy cơ sinh con 47,XXY. Tất cả 47,XXY đều vô sinh.
Phương pháp điều trị
Việc phát hiện bệnh sớm rất hữu ích, mặc dù hiếm khi được chẩn đoán trước tuổi dậy thì. Điều trị cần tập trung vào ba khía cạnh: thiểu năng sinh dục, vú to, và các vấn đề tâm lý xã hội. Trị liệu androgen là yếu tố quan trọng nhất trong quá trình điều trị. Thay thế testosterone nên bắt đầu từ khi dậy thì để bù đắp thiếu hụt androgen, mang lại nét nam tính phù hợp, và cải thiện tình trạng tâm lý xã hội. Tiêm testosterone đều đặn giúp tăng cường sức mạnh, mọc lông mặt, tạo cơ bắp, tăng ham muốn tình dục, tăng kích thước tinh hoàn, cải thiện tâm trạng, hình ảnh bản thân và hành vi, và ngăn ngừa loãng xương sớm. Tiếp cận đa ngành giúp giải quyết rối loạn ngôn ngữ, khó khăn trong học tập và các vấn đề tâm lý xã hội khác.
Phẫu thuật có thể cần thiết để điều trị vú to, vì tình trạng này gây căng thẳng tâm lý xã hội đáng kể và tăng nguy cơ ung thư vú.
Tư vấn di truyền cho thấy tỷ lệ tái phát không cao hơn mức trong dân số chung. Thời điểm tốt nhất để thông báo cho người bệnh là vào giữa đến cuối tuổi thanh niên, khi anh ta đã đủ trưởng thành để hiểu rõ về tình trạng bệnh của mình.
Biến chứng
- Nguy cơ ung thư vú ở nam giới XXY cao gấp 20 lần so với nam giới khỏe mạnh. Các loại ung thư khác xuất hiện ở 1,6% bệnh nhân, bao gồm bạch cầu cấp, lymphoma Hodgkin và không Hodgkin, bạch cầu dòng tủy mạn và các bệnh tăng sinh tủy khác. U tế bào mầm sinh dục và ngoài sinh dục (u tế bào mầm trung thất, u quái, carcinoma quái, carcinoma đệm nuôi) cũng có thể xảy ra.
- Các biến chứng tâm lý và tâm thần có thể xuất hiện ở những người có trí tuệ dưới trung bình, thiểu năng sinh dục, hoặc bất lực.
- Sụp đốt sống có thể xảy ra do loãng xương.
- Phát triển giãn tĩnh mạch và loét chân do ứ đọng tĩnh mạch.
- Các bệnh nội tiết liên quan bao gồm đái tháo đường, suy giáp, hội chứng hố yên rỗng, suy cận giáp, dậy thì sớm liên quan đến u tế bào mầm sản xuất hCG.
- Tăng sinh tuyến tiền liệt lành tính có thể xảy ra do cung cấp testosterone. Bệnh nhân điều trị bằng testosterone nên được tầm soát phì đại tuyến tiền liệt từ tuổi 30.
- Ở nam giới đa nhiễm X, tỷ lệ tử vong do bệnh mạch máu não như bệnh van động mạch chủ và vỡ phình mạch mọng cao hơn 6 lần so với nam giới 25-84 tuổi. Tăng kết tập tiểu cầu, bệnh huyết khối và tăng đông máu đã được phát hiện và có lẽ liên quan đến nồng độ estrogen tăng.
- Mặc dù nhiễm sắc thể Y xác định giới tính nam, nhưng người mắc hội chứng Klinefelter có thể gặp rối loạn xác định giới tính. Tuy nhiên, quan sát này chủ yếu dựa trên báo cáo của các nhóm ủng hộ chuyển giới mà chưa có nghiên cứu khoa học cụ thể.
Tiên lượng
- Các nghiên cứu ban đầu về nam giới hội chứng Klinefelter XXY cho thấy họ tăng nguy cơ rối loạn tâm thần, phạm tội và chậm phát triển trí tuệ. Tuy nhiên, điều này bị nghi ngờ vì những người được thống kê là từ những người đã nhập viện.
- Trẻ nhỏ XXY hầu như không khác biệt so với trẻ khác.
- Dù trẻ trai 47,XXY có thể gặp khó khăn trong học tập, nhiều thất bại, và, trong một số trường hợp, khó khăn nghiêm trọng về hành vi và cảm xúc, nhưng hầu hết đều đạt được sự tự lập đầy đủ khi trưởng thành. Một số hoàn thành giáo dục đại học và có khả năng làm việc bình thường.
- Tuổi thọ được xem là bình thường.
- Thiểu năng sinh dục, khoái cảm tình dục thấp và các vấn đề tâm lý xã hội có thể được điều trị bằng testosterone.
- Vú to có thể được phẫu thuật điều chỉnh.
Biến thể
Hội chứng nam 48,XXYY xảy ra ở một trong 17.000 ca sinh. Truyền thống xem nó là biến thể của hội chứng Klinefelter, nhưng hiện nay nó được coi là một thực thể lâm sàng và di truyền riêng biệt, dù chưa có mã ICD-9. Người mắc bệnh này hung hăng hơn, trí tuệ kém hơn và vóc dáng cao hơn hội chứng Klinefelter.
Hội chứng nam 48,XXXY có chậm phát triển tâm thần nhẹ đến trung bình, chậm nói, phát triển vận động chậm, phối hợp kém, hành vi thiếu trưởng thành, vóc người bình thường hoặc cao, khuôn mặt bất thường (nếp quạt ở mắt, hai mắt xa nhau, môi trề), thiểu năng sinh dục, vú to (33-50%), dương vật kém phát triển, vô sinh, ngón tay lệch, dính xương quay trụ, và điều trị với testosterone có hiệu quả.
Hội chứng 49,XXXYY thường biểu hiện chậm phát triển tâm thần trung bình đến nặng, hành vi thụ động nhưng đôi khi hung hăng và có cơn thịnh nộ, nét mặt biến dạng, vú to, và thiểu năng sinh dục.
Hội chứng nam 49,XXXXY có ba triệu chứng cổ điển là chậm phát triển tâm thần nhẹ đến trung bình, dính xương quay trụ, và thiểu năng sinh dục tăng gonadotropin. Các biểu hiện khác gồm tổn thương ngôn ngữ nặng, rối loạn hành vi, cân nặng lúc sinh thấp, vóc dáng thấp ở một số người, khuôn mặt bất thường (mặt tròn lúc sơ sinh, thô ở tuổi lớn hơn, hai mắt xa nhau, nếp quạt ở mắt, hàm nhô), cổ ngắn hoặc bè, vú to (hiếm), khiếm khuyết tim bẩm sinh (ống thông động mạch thường gặp nhất), bất thường xương (chân vòng kiềng, bàn chân quặp, ngón thứ 5 lệch), giảm trương lực cơ, khớp tăng duỗi, cơ quan sinh dục kém phát triển, và tinh hoàn ẩn. Tinh hoàn kích thước hạt đậu, dương vật nhỏ, và các đặc điểm giới tính thứ phát ấu nhi là đặc trưng ở bệnh nhân 49,XXXXY, trong khi bệnh nhân 48,XXXY biểu hiện thiểu năng sinh dục nhẹ hơn giống 47,XXY.
Nam giới mắc hội chứng Klinefelter có thể có kiểu nhân thể khảm 47,XXY/46,XY, biểu hiện suy giảm sinh tinh ở nhiều mức độ khác nhau. Thể khảm 47,XXY/46,XX có biểu hiện lâm sàng gợi ý hội chứng Klinefelter rất hiếm gặp và cho đến nay chỉ có khoảng 10 trường hợp được báo cáo trong y văn.
Liên kết ngoài
- Viện Sức Khỏe Trẻ Em Quốc Gia
- Hiệp Hội Klinefelter & Các Cộng Sự
- Hiệp Hội Thông Tin & Hỗ Trợ Hội Chứng Klinefelter Hoa Kỳ
- 47xxy.org
- Dữ liệu chi tiết ICD-9-CM 758.7
- klinefeltersyndrome.org
- Nhóm Klinefelter Áo, Thông Tin & Hỗ Trợ Lưu trữ 2016-04-01 tại Wayback Machine
- Hội Chứng XXYY và Dự Án XXYY Lưu trữ 2014-01-07 tại Wayback Machine
- (tiếng Việt) Phát hiện trường hợp Klinefelter ở Việt Nam
Bất thường nhiễm sắc thể | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Nhiễm sắc thể thường |
| ||||||||
| Nhiễm sắc thể giới tính (X/Y) |
| ||||||||
| Chuyển đoạn nhiễm sắc thể |
| ||||||||
| Khác |
| ||||||||